The band theory describes the electronic properties of periodic systems. It calculates the allowed energy states for the electrons within the system. There are two types of bands: the allowed energy bands (valence band & conduction band) and the forbbiden ones (gap). The distribution of this energy bands discriminates materials between, insulators, conductors or semiconductors. To make a band structure calculation, first we have to localize the high symmetry points in the Brillouin Zone of the solid, to study the behaviour of this states.
Figure 1. Brillouin Zone of an fcc structure. Made it on Xcrysden .
Band Structure Calculation
After you have an optimized geometry, cell and ions possition, you can perform a Band Structure calculation.
- First You have to perform a single point calculation with very high accuracy on the SCF (electronic relaxation) to
generate the charge distribution on the system (CHG & CHGCAR file).
- Second You perform a single point calculation using this charge distribution to analize the band structure.
Generate the charge density file
Lets build the charge density of the anatase in volume:
- From a converged geometry use the CONTCAR file as POSCAR
- Use the same POTCAR and KPOINTS file.
- To make a single point calculation to you well known INCAR, change the next instructions:
# Anatase volume #=========================================== # General Setup System = Anatase_benz # Calculation Title PREC = NORMAL # Options: Normal, Medium, High, Low ENCUT = 400 # Kinetic Energy Cutoff in eV ISTART = 0 # Job: 0-new 1-cont 2-samecut ICHARG = 2 # initial charge density: 1-file 2-atom 10-cons 11-DOS ISPIN = 1 # Spin Polarize: 1-No 2-Yes # Electronic Relaxation (SCF) NELM = 150 #Increase the iterations. NELMIN = 2 # Min Number of ESC steps NELMDL = 10 # Number of non-SC at the beginning EDIFF = 1.0E-05 # Increase the stopping criteria for ESC LREAL = .TRUE. # Real space projection IALGO = 48 # Electronic algorithm minimization VOSKOWN = 1 # 1- uses VWN exact correlation ADDGRID = .TRUE. # Improve the grid accuracy # Ionic Relaxation EDIFFG = 1.0E-04 # Stopping criteria for ionic self cons steps NSW = 0 # Max Number of ISC steps: 0- Single Point ISIF = 2 # Ionic Relaxation IBRION = -1 # Single Point ADDGRID = .TRUE. # Improve the grid accuracy SIGMA = 0.10 # Insulators/semiconductors=0.1 metals=0.05 ISMEAR = 0 # Partial Occupancies for each Orbital # -5 DOS, -2 from file, -1 Fermi Smear, 0 Gaussian Smear # Parallelization NPAR=8 NCORE=8
This will generate the charge distribution we will use in the next step
Band Structure
Once you have the CHG files you have to make a new calculation:
- Copy the INCAR POTCAR POSCAR CHG and CHGCAR into the new directory.
We are going to scan the next directions of the Brillouin cell:
Kpoints along high symmetry line 216 line-mode rec 0.0 0.0 0.0 ! gamma 0.0 0.5 0.5 ! X 0.0 0.5 0.5 ! X 0.5 0.5 0.5 ! R 0.5 0.5 0.5 ! R 0.375 0.75 0.375 ! Z 0.375 0.75 0.375 ! Z 0.0 0.0 0.00 ! gamma 0.0 0.0 0.0 ! gamma 0.5 0.5 0.0 ! M
- Now we have to add the next instructions to the INCAR:
- ISTART= 1 Job continuation.
- ICHARG= 11 Job continuation.
- ISIF = 2 Ions relaxation.
- IBRION= -1 Single Point
- NSW = 0 Single Point
- LORBIT= 11 Band Structure
- RWIGS = Wigner Seitz atomic radius
# Anatase Bulk Band Structure #=========================================== # General Setup System = Anatase_benz # Calculation Title PREC = NORMAL # Options: Normal, Medium, High, Low ENCUT = 400 # Kinetic Energy Cutoff in eV ISTART = 1 # Job: 0-new 1-cont 2-samecut ICHARG = 11 # Reads the charge distribution to make a BS ISPIN = 1 # Spin Polarize: 1-No 2-Yes # Electronic Relaxation (SCF) NELM = 150 # More Steps because the criteria NELMIN = 2 # Min Number of ESC steps NELMDL = 10 # Number of non-SC at the beginning EDIFF = 1.0E-04 # Stopping criteria for ESC LREAL = .TRUE. # Real space projection IALGO = 48 # Electronic algorithm minimization VOSKOWN = 1 # 1- uses VWN exact correlation ADDGRID = .TRUE. # Improve the grid accuracy # Ionic Relaxation EDIFFG = 1.0E-02 # Stopping criteria for ionic self cons steps NSW = 0 # Single Point IBRION = -1 # Single Point ISIF = 2 # Stress and Relaxation: 2-Ion 3-cell+ion ADDGRID = .TRUE. # Improve the grid accuracy #BS related values: RWIGS = 1.217 0.820 # Wigner Seits Radius: Ti O LORBIT = 11 # Split the bands SIGMA = 0.10 # Insulators/semiconductors=0.1 metals=0.05 ISMEAR = 0 # Partial Occupancies for each Orbital # -5 DOS, -2 from file, -1 Fermi Smear, 0 Gaussian Smear # Parallelization NPAR=1 NCORE=1
Visualizing the Band Structure
To visualize the Band Structure use p4v software. Download the vasprun.xml file into your computer disk, and excecute p4v. There you will find the tools to plot a DOS and the BS
By the end you must have this plot
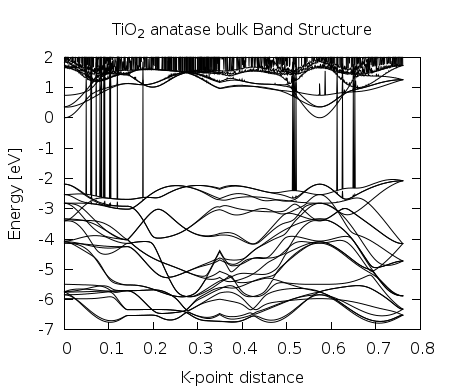
Figure 2. Band Structure generated by Texas utilities and Gnuplot.
You can download the build files here:
INCAR
POTCAR
KPOINTS
POSCAR